临床试验数据质量管理
一、高质量数据集
临床数据管理(CDM)的目标在于,提供一个准确、可靠的高质量可用于分析的数据集,具体来说需满足以下要求:
数据错误需尽可能少,使其能最大限度支持临床试验得出的结论。并不要求无差错数据(error-free),但要求其数据质量能和无差错数据得出一样的分析结论
数据可溯源,亦即监管部门也能利用原始数据得到和申办者相同的分析结论。源数据核查确认SDV和EDC系统的稽查轨迹是保证“可溯源性”的具体手段
数据管理应当有良好保密性,能保护受试者个人信息
对于盲态试验,需保证数据盲态
良好的临床研究数据还应符合ALCOA +CCEA
可归因性 Attributable
易读性 Legible
同时性 Contemporaneous
原始性 Original
准确性 Original
完整性 Complete
一致性 Consistent
持久性 Enduring
可用性 Available
二、数据管理文件
数据管理计划DMP
明确数据管理主要内容及其负责人员和主要时间节点,可用于核查临床试验数据管理的执行情况
数据核查计划DVP
设置若干核查点确保数据的完整性、一致性和合理性,包括但不限于核查是否准确完整录入源数据,是否偏离试验方案,是否随机化实验,是否保持数据间一致性(SAE数据库与临床数据库一致性/外部数据与CRF一致性)等
三、数据收集形式
CRF/eCRF是用来记录受试者临床试验全部信息的纸质或电子文件,需包含研究方案规定的所有收集信息,建议采用CDISC CDASH标准进行设计。CDASH划定了干预、事件、发现三类通用观测分类,又规定了变量名、提示、问题描述文字、数据类型、STDM映像关系,有无codelist等,还给出了每个变量的推荐使用意见“推荐使用HR”,“推荐R/C”,“可选O”
相关关联文件主要有CRF填写指南(CCG)和CRF注释(aCRF)。CCG是指导CRC数据录入的质量文件;aCRF是CRF字段信息和数据库变量映射关系的具体描述,是数据库和CRF间的联系纽带
四、数据存储
数据库设计
电子采集系统EDC用于eCRF的数据收集,但数据存储、医学编码依赖于与EDC集成的临床试验数据管理系统CDMS或临床试验管理系统CTMS。常见的EDC系统有甲骨文公司的OC/RDC和inform,达索公司的MediData Rave(Saas)
CDISC SDTM是NMPA推荐的原始数据库递交格式,其同样包含干预、事件、发现三类通用观测数据集,可与CDISC CDASH建立良好的映像关系。SDTM规定了每个变量的名称、类型、角色等基本属性,虽然不允许添加新变量,但用户可以利用补充修饰数据SUPP设立非标准变量
计算机系统验证
初步设计完成的EDC/CDMS还需进行计算机系统验证,进行计算机系统的质量核查。系统验证旨在证明从设计开始到停止使用,或转换到其他系统的全生命周期均能符合特定要求。GAMP5 是常用的基于风险的计算机验证方法。有关计算机系统在临床试验中的应用,还可参照FDA相关指南和美国21号联邦法规第11部分(CFR 21 Part 11),使得电子签名和电子记录具有和纸质签名和记录相同的法律效力
五、数据输入
研究中心数据
对于研究中心数据,主要是病例记录和本地实验室检查数据,数据录入即是将源数据转录到EDC系,CRC需进行录入前检查,确保录入质量
外部数据
对于研究中心和数据管理机构以外的第三方基地产生的数据,如中心实验室数据、ECG数据、eCOA/ePRO等,采用电子化传输和批加载传输数据至CDMS
六、数据审核与清理
源数据核查SDV
SDV是指CRF数据和源数据间的一致性核查。CRF是所有数据分析的源头,也是经验证明了的错误高频发生所在。CRA需开展基于风险的针对性的源数据核查SDV,确保源头数据的及时准确
源数据审查SDR
与SDV关注源数据本身不同,源数据审查SDR侧重源数据的产生过程。检查其试验实施是否符合试验方案,是否满足依从性要求
逻辑核查
逻辑核查(Edit Check)是计算机系统自动判断录入数据与其预期的数值范围、数值属性间是否存在问题。EDC系统能在数据录入同时实时逻辑核查,极大地提高了eCRF的数据质量。但需注意,对于涉及两个及以上CRF数据的交叉/关联检查,特别是跨访视的变量比较,只能定时统一检查。逻辑核查也只能进行简单的完整性、一致性和合理性检查
人工质量核查
人工核查针对那些语义复杂的数据差异(discrepancy),如。对于逻辑简单的数据偏差,数据管理员DM可进行自明修正SEC,无需发送质疑Query/质疑表DCF给研究者;需要发送质疑的,DM根据答复记录变更数据;特殊情况还需要CRA的协助
医学审查
除却一般的数据质量核查,还需从医学角度进行专业核查,如既往病史HO、伴随用药CM、体格检查PE、入排标准IE间的一致性等,及时发现潜在的安全性风险
汇总分析
借助统计分析工具和一体化数据平台,实时总览临床试验整体质量,比较不同试验中心数据一致性,发现其他核查方式所不能的更隐秘的错误类型,如方案偏离PD、研究者行为不当等,也给CRA的现场监查提供针对性方向
七、AE管理
SAE/SUSAR快速报告
已在其他文章详述
SAE一致性检查
一个SAE既要按CRF要求录入临床试验数据库,又要按快速报告要求上报至安全性数据库。考虑到快速报告的时限要求,部分信息往往缺失,而CRF录入的信息更加完整,也有其他一些原因,导致两个数据库可能并不一致。SAE一致性检查检查两个数据库间关键安全性信息是否一致,而一些微小差异是可以接受的,如描述术语的轻微差异。
数据收集是动态过程,这种检查需在研究中开展多次,数据管理机构有SOP来规定SAE一致性检查的时间间隔
八、质量改进
- 实时关注关键指标和非关键指标错误率,与预设的可接受限值比较。针对高错误率问题进行根本原因分析。若有必要,对SOP或其他质量文件进行修改,或制定纠正和预防措施CAPA
九、数据库最终锁定
- 研究团队收集好所有CRF,解决完所有质疑,完成所有“数据库锁定清单”的要求内容,即可锁定数据库。数据锁定是临床研究的重要里程碑,是指删除访问数据库的路径,防止对数据库文档的无意或未经授权的更改
- 数据库最终锁定后若发现数据错误,如该错误对临床试验的安全性和有效性分析影响有限。部分申办者或选择不改正错误,只将其记录在统计分析报告SAR和临床报告文档CSR中。这需要事先申明的程序来确定哪些数据错误是可以被容忍/无需修改的
- 需谨慎决定重开锁问题,由于数据库锁定后往往紧跟统计分析,重开锁过程极易令人怀疑其真实动机
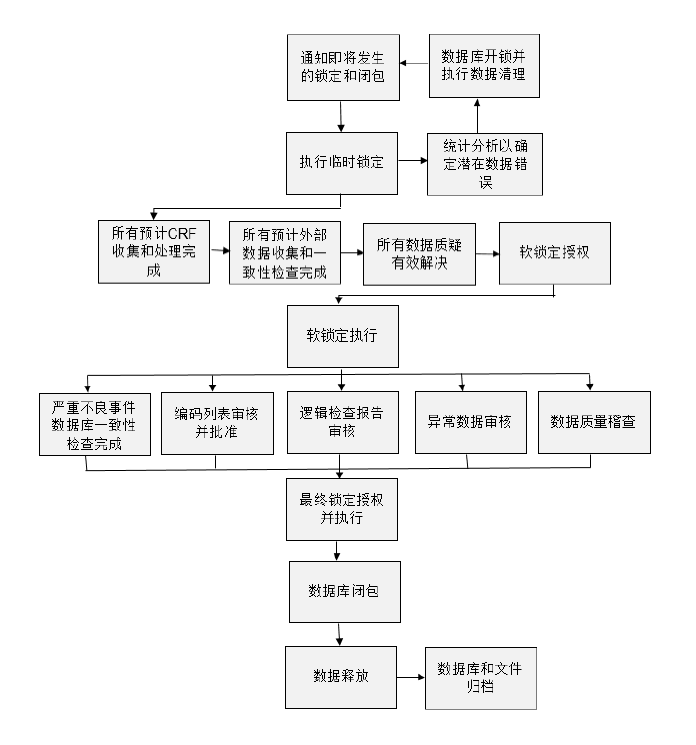
十、统计分析与结果报告
- 数据库最终锁定⋙一次揭盲⋙统计分析⋙二次揭盲⋙结果报告
十一、数据递交
- 根据必备文件要求整理TMF文件夹,根据电子通用技术文档eCTD要求的文件夹结构档归档各类受试者文件,研究文件和支持性文件
十二、文件保存
- 注册用药物临床试验的必备文件保存至少为试验药物被批准上市后 5 年;非注册药物临床试验,则需至少保存至临床试验终止后 5 年
参考资料:
[1] 医药临床研究中的数据管理(颜崇超)
[2] 药物临床试验数据管理工作技术指南
[3] 药物临床试验质量管理规范(2020年版)
[4]药物临床试验数据递交指南原则
[5]药物临床试验数据递交指导原则(试行)
[6] 临床数据质量管理规范GCDMP