SUSAR 快速报告
一、相关术语
不良事件AE
受试者接受试验用药品后出现的所有不良医学事件,表现为症状体征,疾病,或实验室检查异常,不一定与试验用药品有因果关系
严重不良事件SAE
受试者接受试验用药品后出现的严重不良医学事件,具体指:导致死亡、危及生命、永久或严重残疾或功能损伤、导致住院治疗或延长住院时间、先天性异常及出生缺陷、其他重要的医学事件
药物不良反应ADR
与试验用药品可能有关的对人体有害或非预期的反应,该不良事件与试验用药品不能排除相关性,且在临床试验语境下不考虑用药剂量
可疑且非预期严重不良反应SUSAR
性质、严重程度、后果或发生频率不同于有关的源文件(研究者手册,或上市后说明书)所述的严重不良反应,“非预期“,”相关性“,”严重性“缺一不可
治疗期间出现的不良事件TEAE
治疗过程中出现的,且治疗前未出现或相对于治疗前恶化的不良事件,3级TEAE指严重或者具重要医学意义但不会立即危及生命的不良医学事件
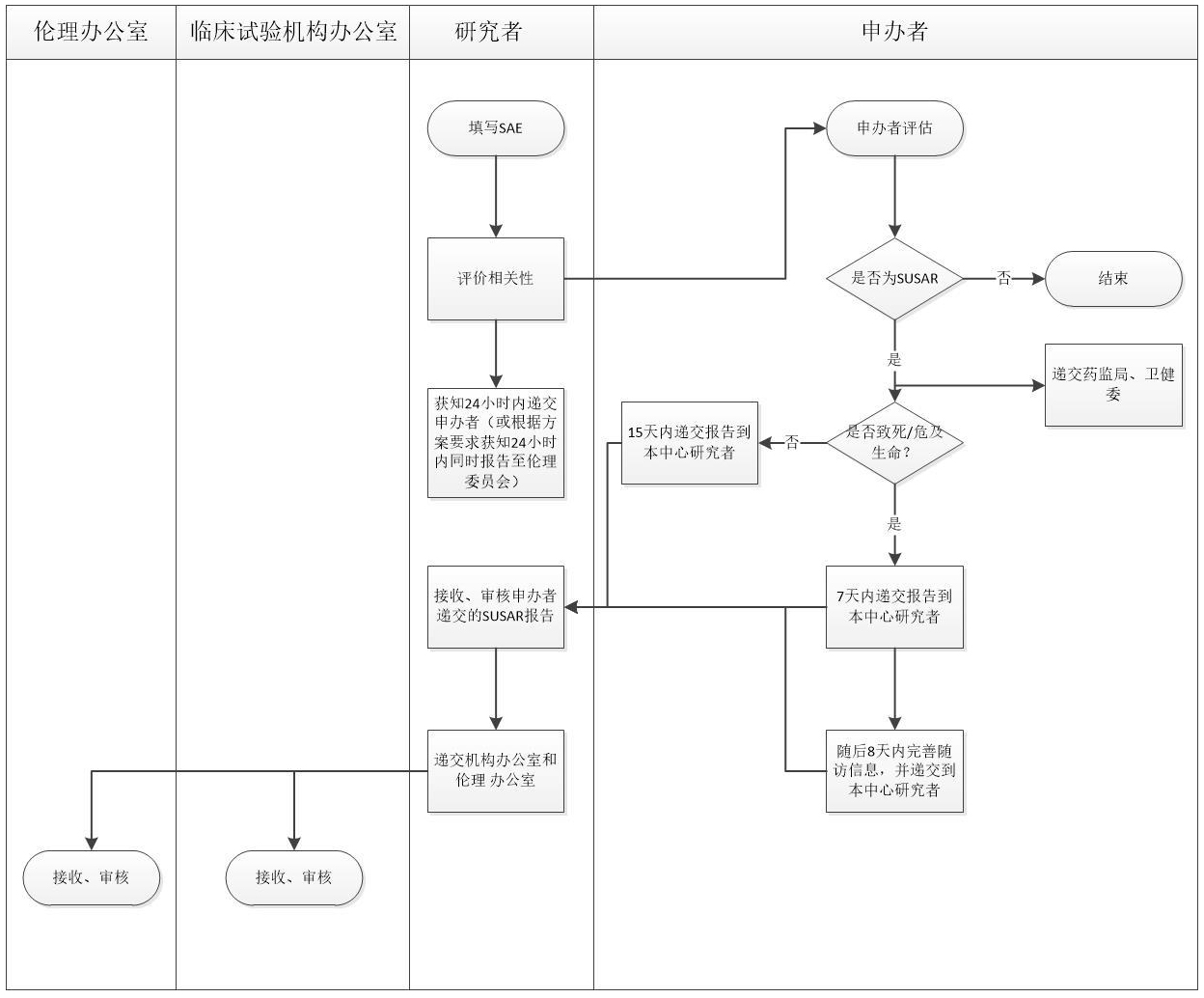
二、SUSAR报告流程
研究者需在获知SAE的24小时内向申办者提交书面报告,并初步评估相关性,除试验方案或者其他文件(如研究者手册)中规定不需要立即上报的。研究者认为需立即向伦理委员会报告的、或根据临床试验方案需上报的 SAE,也需在 24 小时内上报伦理委员会
申报者对SAE进行全面评估和判断,包括严重性、与试验药物的相关性以及是否为预期事件等,将符合SUSAR 定义的报送所有的试验机构、伦理委员会、药监局和卫健委。对于致死或危及生命的SUSAR,申报者在首次获知后不晚于7天(获知当天为第0天)告知国家药品审评机构,在随后的 8 日内完善随访信息并报告;其他SUSAR不晚于15天
研究者需及时阅读签收,并在24小时内向本中心临床试验机构和伦理委员会递交申办者提供的SUSAR报告
三、时限要求
- 首次报告:对于致死或危及生命的SUSAR,申报者在首次获知后不晚于7天(获知当天为第0天)告知国家药品审评机构,在随后的 8 日内完善随访信息并报告;其他SUSAR不晚于15天
- 随访报告:申请人需继续跟踪SUSAR,以随访报告的形式及时报送有关新信息或对前次报告的更改信息等,报告时限为获得新信息起15天内
- 报告时间段:快速报告开始时间为临床试验批准日期/国家药品审评机构默示许可开始日期,结束时间为国内最后一例受试者随访结束日期。临床试验结束或随访结束后至获得审评审批结论前发生的严重不良事件,也应进行快速报告
四、报告形式
- 符合 ICH E2B(R3)的个例安全性报告ICSR形式
五、盲态试验下的快速报告
- 个别病例的破盲不影响整体临床试验数据的最终分析,且可保持其他工作人员如生物统计人员的盲态。如需确定非预期的SAE是否为SUSAR,可进行个例揭盲程序
参考资料:
[1] 药物临床试验期间安全性数据快速报告的标准和程序
[2] 北京大学肿瘤医院医学伦理委员会关于临床试验安全性事件报告的流程与要求
[3] 药物临床试验 安全评价·广东共识(2020年版)
[4] ICH-E2A: 临床安全性数据管理:快速报告的定义和标准
[5] 药物临床试验质量管理规范(2020年版)